心筋代謝
テーマ
心不全における細胞骨格修飾および細胞質代謝変化を介したミトコンドリア機能制御機構の解明
研究背景
心不全は心機能低下として顕在化しますが、その発症・進展には心筋細胞内における構造的変化やエネルギー代謝異常が深く関与しています。特に不全心筋では、ミトコンドリアの酸化的リン酸化低下と解糖系依存の亢進を特徴とする代謝リモデリングが生じますが、その分子レベルの機序は十分に解明されていません。
これまでの研究から、心不全では微小管をはじめとする細胞骨格の構造変化とともに、糖・脂質・アミノ酸代謝に加えて酢酸代謝を含む細胞質代謝の再構築が生じることが示唆されています。私たちは、これらの細胞内変化がミトコンドリア機能障害の単なる結果ではなく、その形成過程に能動的に関与している可能性に注目しています。本研究では、培養心筋細胞および心不全動物モデルを用い、細胞骨格動態および代謝制御の変化がミトコンドリア機能に与える影響を統合的に解析しています。
細胞骨格修飾によるミトコンドリア制御
α-tubulinの脱チロシン化は、心不全心筋で亢進すると報告されています。α-tubulinは微小管を構成し、その翻訳後修飾は細胞内機能制御に重要です。TubulinのC末端構造はミトコンドリア外膜のVDAC(voltage-dependent anion channel)と相互作用し、ミトコンドリア機能の調節に関与します。
私たちは、α-tubulin脱チロシン化をモデル化するために以下の細胞系を構築しました。
・Vasohibin(VASH)過剰発現H9c2細胞
・Tubulin tyrosine ligase(TTL)ノックアウトH9c2細胞
これらの細胞では、ミトコンドリア呼吸能の低下や膜電位の減弱がみられ、かつミトファジー初期過程で重要なVDACのポリユビキチン化が低下していました。これらの結果は、脱チロシン化α-tubulinが劣化ミトコンドリアの識別・除去を阻害し、マイトファジー低下を介してミトコンドリア機能を悪化させる可能性を示しています。
さらに、心筋特異的VASH過剰発現マウスでは、HFpEF様表現型および運動耐容能低下が観察され、この細胞骨格修飾が心機能にも影響を与えることが示されました。
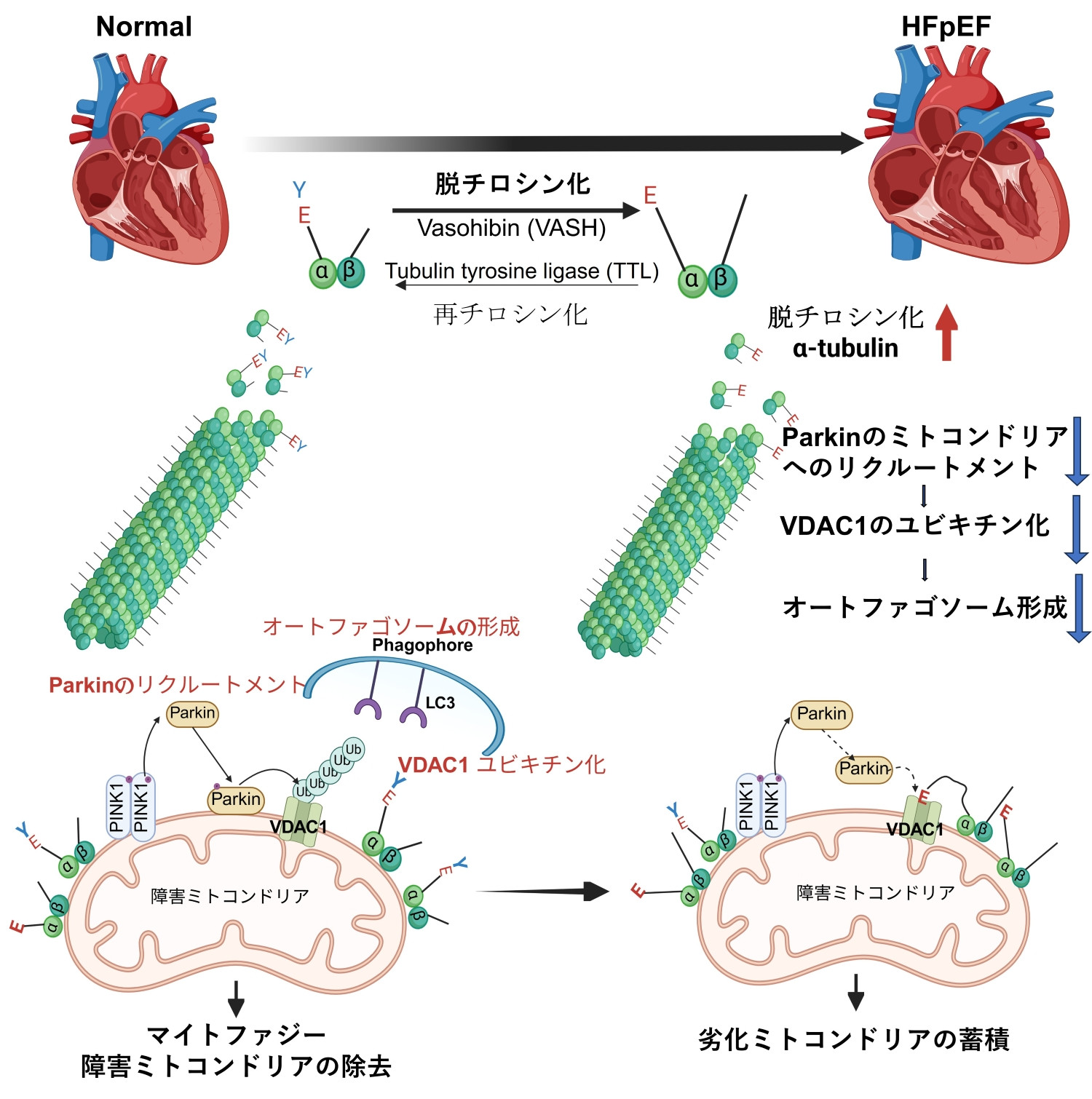
脱チロシン化α-tubulinの増加を介した心機能低下の機序
細胞質代謝(酢酸–アセチルCoA)とミトコンドリア機能
心不全における代謝リモデリングでは、細胞質アセチルCoA恒常性の破綻も重要な要素として浮かび上がっています。慢性的なβアドレナリン刺激下では、細胞質内でアセチルCoAを産生する主要酵素であるACSS2(acetyl-CoA synthetase short-chain family member 2)の発現が心筋で低下することを見出しました。
ACSS2の低下は細胞質アセチルCoAの枯渇を引き起こし、ミトコンドリア外膜VDACのアセチル化低下および不安定化を介して、ミトコンドリアの形態変化や酸化的リン酸化能低下をもたらしました。一方で、ACSS2を過剰発現させるとミトコンドリア構造・機能は保たれ、β刺激による心機能低下も有意に抑制されました。
これらの知見は、ACSS2を介した細胞質アセチルCoA恒常性の破綻が、VDACを中心としたミトコンドリア品質制御異常を引き起こす新たな病態機構である可能性を示しています。
主な業績
- 1.Miura S, Misaka T, Sekine T, Ogawara R, Ichimura S, Tomita Y, Yokokawa T, Oikawa M, Ishida T, Takeishi Y. Detyrosinated α-tubulin mediates mitochondrial dysfunction and diastolic impairment in heart failure with preserved ejection fraction. FEBS Lett. 2025; 599: 2474-2490.
DOI: 10.1002/1873-3468.70119
文責:三浦俊輔