

The most common phenotypes of idiopathic inflammatory myopathies (IIMs) include polymyositis, dermatomyositis, immune -mediated necrotizing myopathy (IMNM), and inclusion body myositis1,2). The detection of myositis-specific autoantibodies (MSAs) led to the reclassification of IIM to account for this antibody group3,4). Anti-signal recognition particle (SRP) antibodies are found in 4%–6% of patients with IIM and are associated with severe necrotizing myopathy despite the absence of inflammatory infiltrates on biopsy5,6). The average age at onset of anti-SRP antibody-positive myositis is 36–52 years, with a slight predominance in females7,8). The disease is clinically limited to muscle symptoms, with a lack of systemic involvement of the skin, lungs, and other body parts, and few complications associated with malignancy or other collagen diseases. The myopathy observed in this disease is severe, and patients present with high serum creatine kinase (CK) levels5,8). Anti-SPR antibody-positive myositis is often steroid-resistant, and additional immunosuppressive agents are sometimes required. The efficacy of various immunosuppressive agents, including tacrolimus, rituximab(RTX), methotrexate, azathioprine, mycophenolate mofetil, and cyclosporine, has been reported5,6,9). However, owing to its rarity and the limited number of case reports on the disease, there is no established treatment regimen for anti-SPR antibody-positive myositis. Herein, we describe the case of a 51-year-old man with anti-SRP antibody-positive myositis refractory to high-dose steroids and a classical immunosuppressive drug, with symptom improvement following treatment with intravenous cyclophosphamide (IV-CY).
The content of research paper
Successful treatment of anti-signal recognition particle antibody-positive myositis with intravenous cyclophosphamide: A case report
Jumpei Temmoku, Shuhei Yoshida, Kanae Tsuchihashi, Yuya Sumichika, Kenji Saito, Haruki Matsumoto, Yuya Fujita, Naoki Matsuoka, Tomoyuki Asano, Nozomu Matsuda, Shuzo Sato, Kiyoshi Migita
-
Jumpei Temmoku
Department of Rheumatology, Fukushima Medical University School of Medicine
-
Shuhei Yoshida
Department of Rheumatology, Fukushima Medical University School of Medicine
-
Kanae Tsuchihashi
Department of Neurology, Fukushima Medical University School of Medicine
-
Yuya Sumichika
Department of Rheumatology, Fukushima Medical University School of Medicine
-
Kenji Saito
Department of Rheumatology, Fukushima Medical University School of Medicine
-
Haruki Matsumoto
Department of Rheumatology, Fukushima Medical University School of Medicine
-
Yuya Fujita
Department of Rheumatology, Fukushima Medical University School of Medicine
-
Naoki Matsuoka
Department of Rheumatology, Fukushima Medical University School of Medicine
-
Tomoyuki Asano
Department of Rheumatology, Fukushima Medical University School of Medicine
-
Nozomu Matsuda
Department of Neurology, Fukushima Medical University School of Medicine
-
Shuzo Sato
Department of Rheumatology, Fukushima Medical University School of Medicine
-
Kiyoshi Migita
Department of Rheumatology, Fukushima Medical University School of Medicine
Introduction
Case Report
A 51-year-old man presenting with weight loss, proximal muscle weakness, and dysphagia was admitted to a local hospital. The patient had no history of neurological or autoimmune disease. He had a 25-year history of smoking 20 cigarettes per day from 20 to 45 years of age. Two years before admission, the patient started experiencing bilateral difficulties raising his upper limbs. One month before admission, he started experiencing lumbar pain and bilateral weakness in his thighs. Over 2 months, he lost approximately 15 kg of body weight, for which he was examined by his local doctor and admitted to the hospital. Skeletal muscle magnetic resonance imaging (MRI) revealed high signals on short inversion time inversion recovery (STIR) in the trunk and proximal upper and lower limb muscles (Figure 1). Needle electromyography revealed denervation potentials and myogenic changes, and myositis was suspected. Subsequently, the patient was transferred to our hospital. Upon admission, the patient’s vital signs were as follows: temperature, 36.6°C; heart rate, 98 beats/minute; and blood pressure, 137/89 mmHg. Physical examination revealed no systemic skin rash, arthralgias, or edema; no cranial nerve abnormalities were noted. The patient’s grip strength was 22.5 kg and 16.5 kg in the right and left hands, respectively. The manual muscle strength test revealed proximal muscle weakness; neck flexion, deltoid, iliopsoas, and gluteus medius muscle strengths were 2/2, 2/2, 2/2, and 4/4, respectively. The patient had normal deep tendon reflexes in all four limbs and no ataxia or sensory disturbances. The patient could stand and walk independently but had claudication with pelvic sway while walking. Laboratory findings revealed elevated serum levels of CK at 10,093 U/L, lactate dehydrogenase level of 1,264 U/L, and aldolase level of 217 U/L (Table 1). Anti-nuclear, anti-ribonucleoproteins, anti-Smith, anti-Jo-1, anti-melanoma differentiation-associated protein 5, anti-aminoacyl tRNA synthetase, antiMi-2, and anti-transcriptional intermediary factor 1γ antibodies were negative. Anti-SRP antibodies were positive with titers of 3.5 IU/mL. Electrocardiography revealed no abnormalities. Respiratory function tests revealed a vital capacity of 68% and a percent forced expiratory volume in one second of 70.5%, indicating restrictive ventilatory impairment. Tumor markers (carcinoembryonic antigen, carbohydrate antigen 19-9, and cancer antigen 125) were negative, and contrast-enhanced computed tomography demonstrated no findings indicative of malignancy. A muscle biopsy of the left biceps brachii was performed to confirm the diagnosis. Histological examination revealed irregular muscle fiber sizes, and scattered necrotic and regenerating fibers, but no perimysial atrophy, intramysial sheath fibrosis, or mononuclear cell infiltration (Figure 2A). Immunohistological staining revealed mild, but diffuse, human leukocyte antigen-ABC expression on myofiber membranes (Figure 2B) and deposition of the membrane invasion complex, the complement C5b-9 complex, on the myofiber membrane in one muscle fiber (Figure 2C). Type 2C fibers were scattered (Figure 2D), and myofibers with diffuse cytoplasmic p62-positive fine aggregates were observed (Figure 2E). A diagnosis of anti-SRP antibody-positive IMNM was made based on the European Neuromuscular Centre classification9). The patient’s clinical course is detailed in Figure 3. Corticosteroid pulse therapy (methylprednisolone 1,000 mg/day) was administered intravenously for 3 days starting on day 6 of admission, followed by 50 mg/day (1.0 mg/kg body weight) of oral prednisolone. Subsequently, corticosteroid pulse therapy was re-administered owing to myalgia symptoms and no myogenic enzyme improvement on biochemical tests on day 13 of admission. Tacrolimus (3.0 mg/day) was administered orally from day 20, and intravenous immunoglobulin was administered for 5 days from day 26. Despite receiving high doses of glucocorticoids and tacrolimus, the patient’s CK levels remained elevated, and he developed dyspnea as well as dysphagia. MRI imaging on day 46 revealed high STIR signals, and we determined that the myositis was still active. IV-CY (750 mg) was administered every 4 weeks starting on day 48 to reduce the prednisolone dose and control the myositis. After treatment initiation, muscle weakness improved from day 76, and myogenic enzyme levels decreased further. Prednisolone was gradually reduced, and the patient was discharged on day 90. He continues to attend the outpatient clinic, where IV-CY has been administered three times, tacrolimus has been reduced to 3.0 mg/day, and prednisolone has been reduced to 9.0 mg/day. On day 44 after discharge from the hospital, blood tests demonstrated CK normalization to 144 U/L, with no symptom flare-ups.
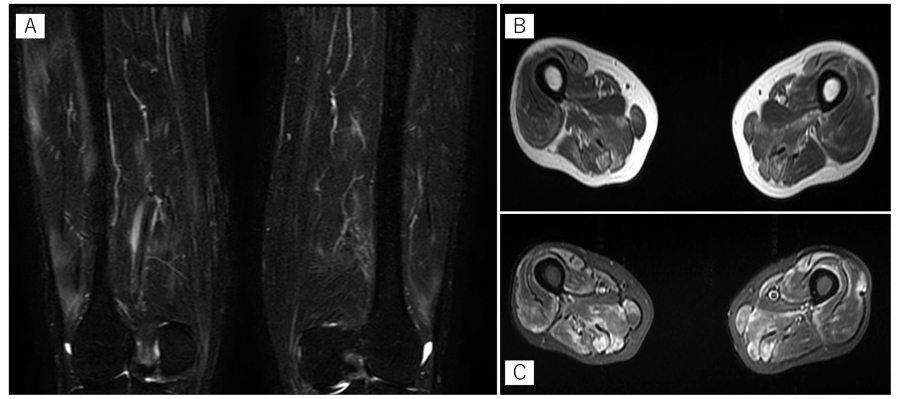
Fig. 1.
MRI findings
MRI revealed a high signal on STIR and T1-weighted images in the upper and lower limb muscles bilaterally. MRI findings suggested muscle inflammation. (A) STIR coronal images of the bilateral lower thighs. (B) T1-weighted bilateral horizontal images of the lower thighs. (C) STIR bilateral horizontal image of the lower thighs. MRI:magnetic resonance imaging, STIR:short inversion time inversion recovery.
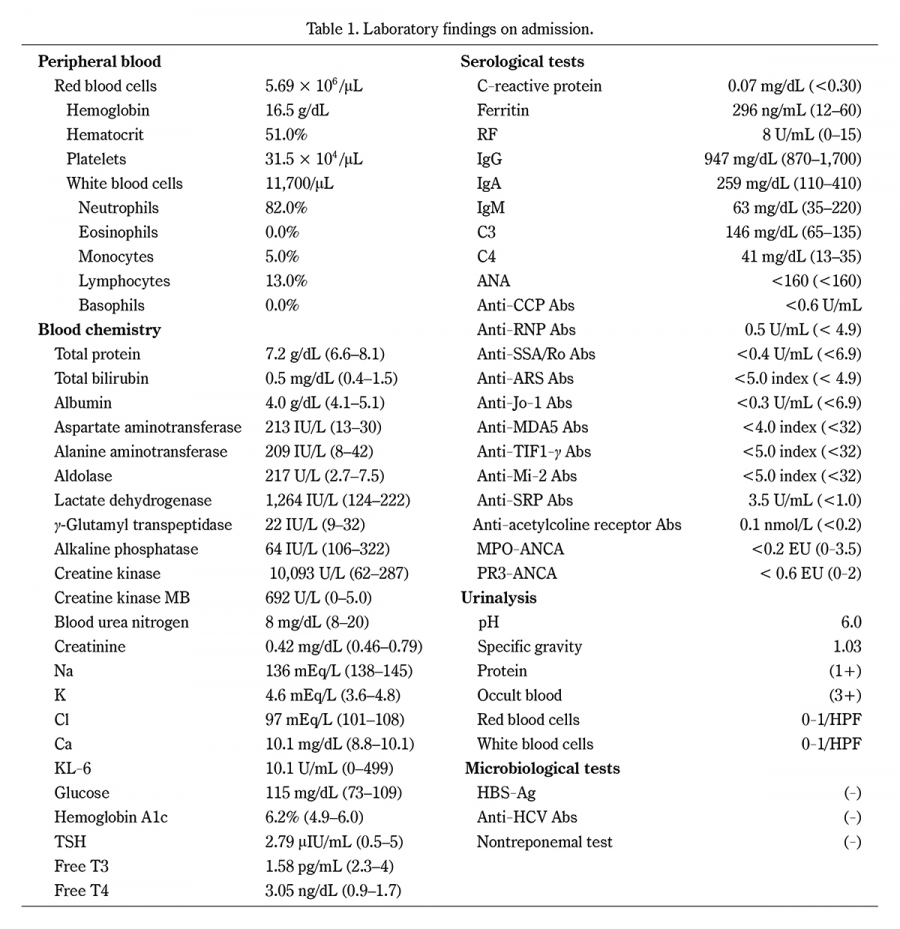
Table 1.
Laboratory findings on admission.
KL-6, Sialylated carbohydrate antigen; TSH, thyroid stimulating hormone; RF, rheumatoid factor; Ig, immunoglobulin; ANA, antinuclear antibodies; CCP, cyclic citrullinated peptide; Abs, antibodies; RNP, ribonucleoprotein; ARS, aminoacyl-tRNA synthetase; MDA5, melanoma differentiation-associated gene 5; TIF1γ, transcription intermediary factor 1-γ; SRP, signal recognition particle; MPO-ANCA, myeloperoxidase-anti-neutrophil cytoplasmic antibodies; PR3-ANCA, proteinase 3-anti-neutrophil cytoplasmic antibodies; HPF, high power field; HBsAg, hepatitis B virus surface antigen; HCV, hepatitis C virus.
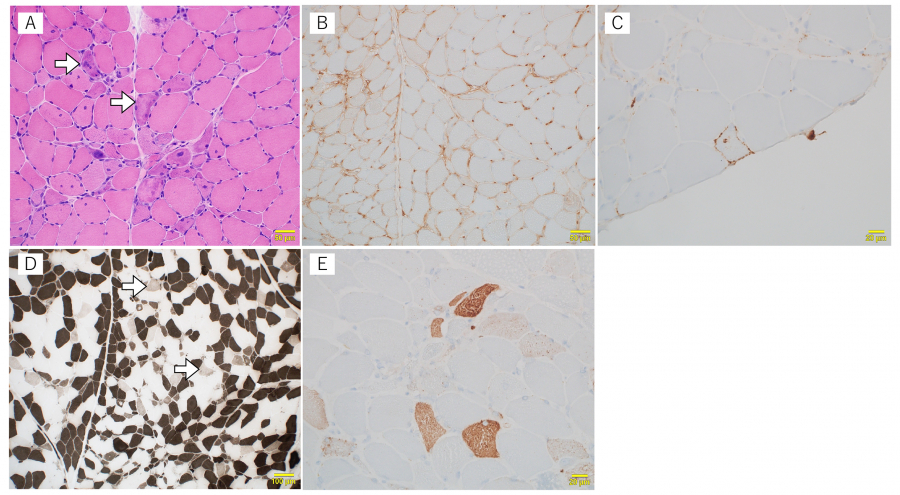
Fig. 2.
Histopathological findings of the muscle biopsy tissue
Hematoxylin and Eosin staining showed increased internal nuclei, numerous necrotic myofibers (arrows), and moderate fiber size variations (A), positive major histocompatibility complex class I (B), and membrane attack complex (C) immunostaining. (D):Type 2C fibers (arrow; fibers of the intermediate color), which reflect immaturity of myofibers, are scattered. This finding suggests the presence of active necrotic and regenerating processes. Adenosine triphosphatase stain at 4.3 pH. Darkly stained fibers are type 1 fibers, while the white ones are type 2A or 2B fibers. (E):Sarcoplasmic p62 aggregates in a fine granular pattern.
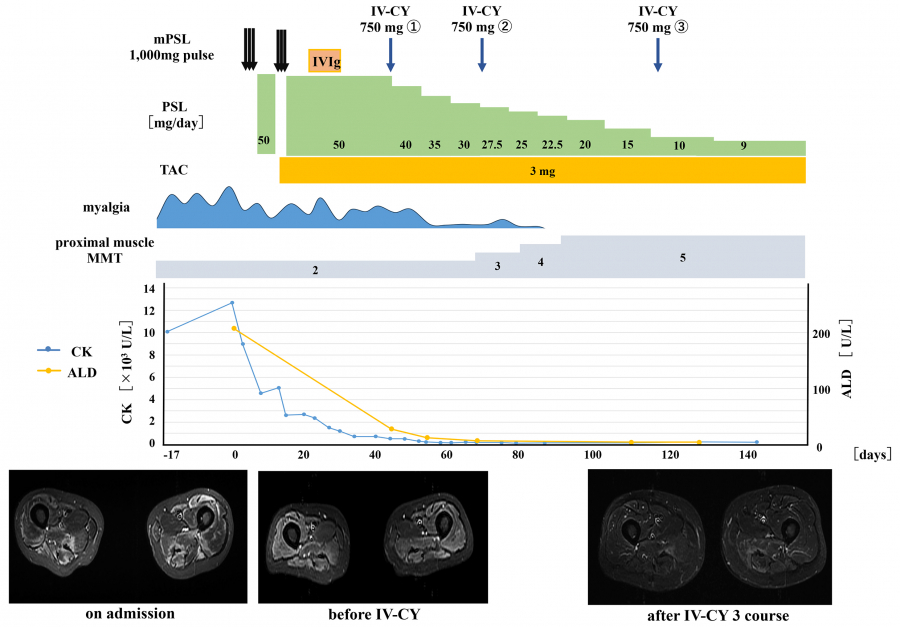
Fig. 3.
The clinical course of the 51-year-old male patient after his IMNM diagnosis
Day 0 indicates the date of hospital admission. After admission, steroid pulse therapy, high-dose gamma globulin therapy, and oral tacrolimus were administered. However, his muscle weakness did not improve. The patient experienced persistent muscle weakness and myalgia, which gradually improved after intravenous cyclophosphamide therapy.
IMNM:immune-mediated necrotizing myopathy.
Discussion
Herein, we presented a case of anti-SRP antibody-positive myositis successfully treated with IV-CY. Although the mechanism of IIM is not fully understood, several histopathological studies have suggested that the condition is a T-cell-mediated disease10,11). However, the presence of MSAs in approximately 40%–90% of patients suggests an ancillary B-cell role12,13). Recently, clinical trials have suggested that RTX, an agent specifically targeting B-cells, may be useful for refractory IIM treatment14,15). This study highlighted that B- and T-cells could be important IIM therapeutic targets. Furthermore, several case reports and series have suggested the efficacy of RTX in treating anti-SRP antibody-positive myositis6,14,16). Valiyil et al. reported the clinical course of eight patients with refractory anti-SRP antibody-associated myositis treated with RTX and found that six patients responded well to RTX treatment. A larger longitudinal study by Pinal-Fernandez et al. found significant clinical improvement in 13 of 17 patients with anti-SRP antibody-positive IIM treated with RTX. Furthermore, this study demonstrated that the RTX treatment benefit was maintained for more than 2 years in many patients16). Therefore, RTX may be a treatment option for refractory anti-SRP antibody-positive myositis6). However, RTX is not currently covered by Japanese medical insurance for myositis treatment, and no large-scale studies have compared RTX efficacy with that of other immunosuppressive agents in anti-SRP antibody-positive myositis. Further studies are required to establish a treatment protocol for anti-SPR antibody-positive myositis.
CY nonspecifically suppresses immune cells, including T-cells, B-cells, and plasma cells, and is effective in various rheumatic diseases, such as myositis, rheumatoid arthritis, systemic lupus erythematosus, and vasculitis17-21). Mecoli et al. reported the clinical course of five patients with refractory IMNM treated with IV-CY (three with anti-SRP antibodies and two with anti-HMGCR antibodies). Of the three patients with positive anti-SRP antibodies, two demonstrated dramatic improvement in muscle strength and muscle enzyme levels following CY treatment and remained stable after treatment22). In our case, the patient did not demonstrate complete symptomatic or radiological improvement after high-dose steroids and tacrolimus treatment. However, the clinical findings improved after IV-CY treatment, and there was no myositis relapse despite early steroid reduction. Thus, the immunosuppressive effect of CY described in this case may have suppressed myositis activity, leading to steroid tapering. Although long-term follow-up is necessary, IV-CY may be an effective treatment option for anti-SRP antibody-positive myositis.
Financial Support
This report was supported partly by an Intramural Research Grant (5, 6) for Neurological and Psychiatric Disorders of the NCNP.